
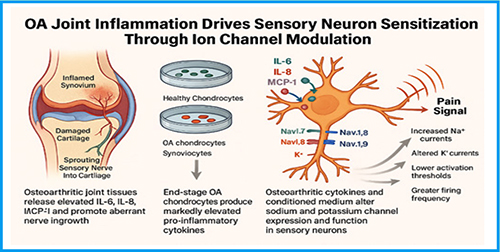
Abstract 087 | Ion channel dysregulation and neuronal sensitization as drivers of osteoarthritis pain
Lukas Weigl 1, Bibiane Steinecker-Frohnwieser 2 | 1Clinical Department of Special Anaesthesia and Paintherapy; 2Medical University of Vienna, Vienna, Austria.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Published: 2 March 2026
Downloads
1. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019 Apr 27;393(10182):1745-1759. Doi: 10.1016/S0140-6736(19)30417-9. DOI: https://doi.org/10.1016/S0140-6736(19)30417-9
2. Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: A review. JAMA. 2021; 325:568-578. doi: 10.1001/jama.2020.22171. DOI: https://doi.org/10.1001/jama.2020.22171
3. Miller RE, Malfait AM. What is new in pain modification in osteoarthritis? Rheumatology. 2018; 57(suppl_4): iv99-iv107. doi: 10.1093/rheumatology/kex522. DOI: https://doi.org/10.1093/rheumatology/kex522
4. Trouvin AP, Perrot S. Pain in osteoarthritis. Implications for optimal management. Joint Bone Spine. 2018 Jul; 85(4):429-434. doi: 10.1016/j.jbspin.2017.08.002. DOI: https://doi.org/10.1016/j.jbspin.2017.08.002
5. Suri S, Walsh DA. Osteochondral alterations in osteoarthritis. Bone. 2012; 51:204-211. doi: 10.1016/j.bone.2011.10.010. DOI: https://doi.org/10.1016/j.bone.2011.10.010
6. Fu W, Vasylyev D, et al. Nav1.7 as a chondrocyte regulator and therapeutic target for osteoarthritis. Nature. 2024; 625: 557–565 (2024). doi: 10.1038/s41586-023-06888-7.
How to Cite

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
PAGEPress has chosen to apply the Creative Commons Attribution NonCommercial 4.0 International License (CC BY-NC 4.0) to all manuscripts to be published.